

 Global
Global  United States
United States 
Navigating the FDA Maze for Smart Medical Devices
Smart medical devices are specialized hardware which are usually coupled with software systems and features such as interoperability, connectivity, power management etc. to perform certain healthcare-related functions such as monitoring and managing patients’ health and accurate real-time diagnosis and medical data analysis in clinical research. Embarking on the journey of medical device design and development services, equipped with microprocessors, displays, and internet connectivity—means navigating a complex web of FDA regulations for medical devices. Think of it as preparing for a space mission where every component must work perfectly! The current medical devices market is expected to achieve a revenue of US $509.9 billion by 2024 and is projected to reach a market volume of US 4673.1 billion by 2029 surging at a CAGR of 5.71%.
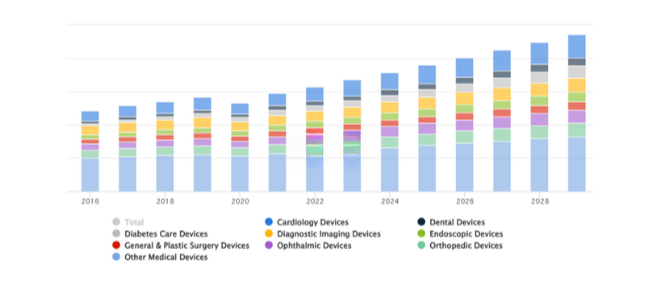
Source: Statista
Increase in development with FDA regulations for medical devices from 2018-2028
Key Guidelines for Launching Your Device
Quality System Regulation (21 CFR Part 820):
- It is the blueprint for building safe and effective devices. It covers everything from design to production and beyond.
- It ensures your device is built exactly as intended and operates safely every time.
- This regulation for medical devices covers the methods and documentation for the design, testing, production, processes, controls, and maintenance of medical devices. It ensures that devices are consistent with their intended design and are safe and effective for their intended use.
General and Special Controls (21 CFR Part 860):
- It throws light on the basic requirements plus extra safety checks for more complex devices.
- General controls include provisions that relate to adulteration; misbranding; device registration and listing; premarket notification; banned devices; notification, including repair, replacement, or refund; records and reports; and good manufacturing practices.
- Special controls may include special labeling requirements, mandatory performance standards, and post-market surveillance.
Software Validation (General Principles of Software Validation; Final Guidance for Industry and FDA Staff):
- It makes sure the software within your device is dependable and up to the task.
- It keeps your device running smoothly, avoiding software hiccups that could affect performance.
- This guidance emphasizes the importance of software validation for devices that include software (as a component, part, or accessory) or are themselves software. This is crucial for ensuring the reliability of software in the functioning of medical devices.
Wireless Compliance (21 CFR Part 15):
- It ensures compatibility with other electronic devices.
- It prevents your device from causing electronic interference, which is crucial in a hospital full of sensitive equipment.
- Devices that include wireless components must comply with FCC’s list of healthcare regulations concerning electromagnetic interference and radio frequency emissions to ensure they do not interfere with other medical equipment.
Medical Device Reporting (MDR, 21 CFR Part 803):
- It reports any problems post-launch to keep tabs on device performance.
- It acts as a continuous feedback loop that helps you make necessary adjustments.
- This requires manufacturers to report adverse events and certain malfunctions to the FDA medical device regulations, which could be particularly relevant for devices with extensive software or electronic components.
HIPAA Compliance and Data Security/Privacy Practices:
- HIPAA Law Compliance:
- It ensures the protection of patient health information (PHI) and compliance with the Health Insurance Portability and Accountability Act (HIPAA) regulations.
- It protects patient privacy and confidentiality, prevents unauthorized access to PHI, and ensures the secure handling of sensitive medical data.
- Compliance with HIPAA regulations are essential for medical devices that handle, store, or transmit PHI, including smart medical devices with internet connectivity or data storage capabilities.
- Data Security and Privacy Practices:
- It includes implementation of robust data security measures and privacy practices to safeguard patient data and mitigate the risk of data breaches or unauthorized access.
- It protects patient confidentiality, maintains data integrity, and builds trust among users and stakeholders.
- Data security and privacy practices include encryption of sensitive data, access controls, regular security audits, secure data transmission protocols, and compliance with industry best practices and standards.
- Medical Device Software Development: IEC 62304
- It is the cornerstone international standard for dependable medical device software lifecycle processes.
- It sets the framework for the entire development lifecycle of medical software, ensuring that each phase—from software design and development to maintenance and risk management—meets stringent safety and reliability standards.
- Adherence to IEC 62304 is crucial, especially for devices that rely heavily on software to operate correctly. This ongoing vigilance helps prevent software failures that could lead to device malfunctions, thereby safeguarding both patient health and manufacturer credibility.

HIPAA compliance best practices & regulations for medical devices
Class 3 Medical Device Deployments
As we are aware, the FDA categorizes embedded medical devices into three types based on risk level posed by them and regulations for medical devices they fall under for patient safety. They are Class 1, which are low risk/harmful devices that do not sustain life and do not need to undergo premarket approval process like stethoscopes and ECG machines, Class 2, which pose higher risk than Class 1, are subjected to regulatory controls and require post-market surveillance and special labeling like catheter, syringes, blood transfusion kits etc., and the final category of Class 3 devices that pose highest risk and support human life such as pacemakers, implants etc. For Class 3 devices—think of these as the high-stakes players due to their critical roles in health:
- Premarket Approval (PMA, 21 CFR Part 814): The FDA’s ‘seal of approval’ process. It’s rigorous but crucial for devices that can have a profound impact on health. This is the FDA medical device regulation process of scientific review to evaluate the safety and effectiveness of Class 3 medical devices. PMA is the most stringent type of device marketing application required by the FDA and includes the submission of clinical data to support claims.
- Clinical Trials: Testing your device in real-world scenarios to ensure it’s both safe and effective. Most Class 3 devices, unless specifically exempted, require clinical trials to gather safety and efficacy data. The FDA regulations for medical devices review this data as part of the PMA process.
- Post-market Surveillance: Keeping an eye on your device even after it’s in use, to ensure long-term safety.
- ISO 13485: This standard of healthcare industry regulations ensures you have the right tools and processes to consistently create medical marvels. It isn’t just about making devices; it’s about ensuring they’re safe enough to be trusted with lives. It sets up shields around the process, protecting both the maker and the user. Adhering to ISO 13485 isn’t just good practice; it’s your passport to international markets. It’s like having a universal translator for meeting global safety standards. It reassures stakeholders, regulatory bodies, and customers that your device is the result of a meticulously managed process focused on top-notch quality and safety. This standard demands ongoing improvement. It’s not a one-time victory but a lifelong commitment to excellence and innovation.
- Risk Management: Adhering to ISO 14971 is needed to anticipate and mitigate risks throughout the device’s lifecycle. While not solely a regulatory code, compliance with ISO 14971, an international standard for the application of risk management to medical devices, is considered best practice and often expected for Class 3 devices.
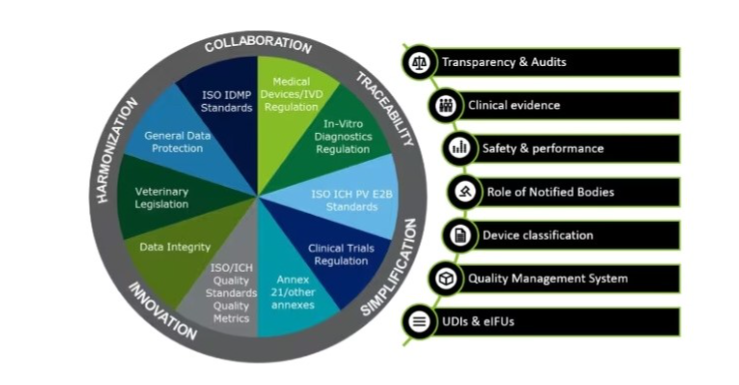
Trends of regulations in healthcare
Trends in Regulatory Guidelines
The regulatory landscape for medical devices is continuously evolving, with recent changes impacting how developers approach minimum viable product vs prototype development. Here are some key regulatory changes and trends:
EU Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR):
The MDR, which replaced the Medical Devices Directive (MDD) in May 2021, introduced stricter requirements for medical device certification in the European Union. It emphasizes a lifecycle approach to safety, increased transparency, and more rigorous post-market surveillance. The IVDR, effective from May 2022, imposes similar stringent requirements on in vitro diagnostic devices.
FDA’s Software as a Medical Device (SaMD) Guidelines:
The FDA medical device regulation has issued updated guidelines for SaMD, which provide clarity on how software functions intended for medical purposes are regulated. The guidelines emphasize the importance of clinical evaluation, real-world performance monitoring, and cybersecurity measures.
Cybersecurity and Data Privacy Regulations:
With the increasing integration of connected devices and IoT-related regulations in healthcare, regulatory bodies are focusing more on cybersecurity and data privacy. The FDA and EU MDR both emphasize the need for robust cybersecurity measures in medical devices.
Real-World Evidence (RWE) in Regulatory Submissions:
Regulatory bodies like the FDA are increasingly accepting real-world evidence as part of the approval process. RWE includes data collected outside of traditional clinical trials, such as from electronic health records (EHRs) and patient registries.
Final Thoughts
The definition of medical devices is expanding daily, and it is the hour’s need to obtain registration for newly developed devices as per Government regulations. Businesses must adopt ISO 13485 and other standards within the stipulated time provided by them for hitherto unregulated medical devices. The responsibility now lies with the industry to reinforce the trust of Indian consumers and the international community in the quality and safety of medical devices sold in the market. They need to establish human-centered design principles and user-friendly features in the development cycle while leveraging emerging technologies that redefine healthcare industry regulations, which can be done by collaborating with industry experts like KritiKal Solutions. With its team of experienced engineers and designers, you can develop advanced versions of medical devices that comply with regulatory standards, navigate the world of medical device development more manageable. Please mail us at sales@kritikalsolutions.com for successful medical device development and market launch which will broaden the horizons of healthcare advancements.

Mohamed Wasif currently works as an Associate Architect at KritiKal Solutions. He is an experienced software developer with a decade of work in C++, C, Linux Kernel, System Design, UML, alongside a proven track record of delivering results, leading teams and optimizing product performance. With his passion for innovation and commitment to continuous learning, he has helped KritiKal in delivering various major projects.