


 Global
Global  United States
United States 

Introduction to Medical Device Design Engineering
A medical device is defined as an instrument, apparatus, machine, appliance, implant, reagent or software which is used for diagnosing, treating or preventing a particular disease or health condition and data analysis in clinical research. It can range from simple devices like laboratory thermometers to complex ones such as pacemakers and surgical systems. Designing and development of medical devices is a multi-faceted journey that needs the expertise of skilled professionals, and governance through standards for safety, efficacy and compliance with relevant medical regulations. The complete process of developing a medical technology solution involves certain crucial steps such as conception, medical product design, development, testing, mass production, regulatory approval and post-launch surveillance.
As of 2024, the medical device market has achieved a market value of approximately US $509.9 billion and is projected to reach a volume of US $4673.1 billion by 2029 increasing at a CAGR of 5.71%. With an increase in manufacturing and accessibility to sophisticated technology across the world, it becomes more necessary to create design methodologies and engineering practices that follow regulatory frameworks effectively. Let us further go through the intricacies of the complex terrain of medical device design process in this blog which may assist those navigating the same with invaluable insights.
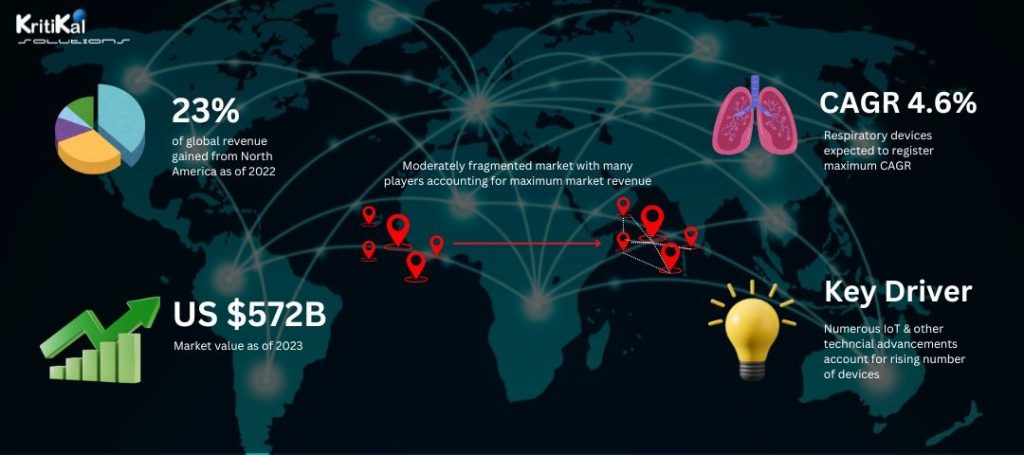
Increasing market size, trends and demographics of medical device design services
Steps of Medical Device Design and Development
Given below is an account of various phases of designing and developing medical devices or healthcare device innovation, where every step needs to be adhered to international regulations, client and patient requirements, understanding of the intended use and risk management procedures.
Medical Device Prototyping
Unmet medical requirements or solutions that need improvement are considered during the starting phase of the designing journey. The medical device market landscape is analyzed for gaps, demands and opportunities, which are later on aligned with user requirements. Once a requirement is recognized and analysis is conducted, the conceptualization and feasibility analysis process are set in place to bridge the gap between the need and the solution. Ultimately, potential use cases of the proposed medical device can assist in making decisions around subsequent phases. A low fidelity mock-up is developed to conduct initial tests and gather early feedback from patients.
Design & Development
It is of utmost importance to ensure that medical device design process follows established control regulations that oversee safety, quality and mitigate patient health-related risks. For example, FDA regulations for medical devices like 21 CFR 820.30 provides guidance for the complete medical device lifecycle including designing, production and distribution. The control process for medical device design for entrepreneurs includes design inputs where client requirements are translated into design needs through repetitive process as per the Safe Medical Devices Act. Through the same, the design inputs are transformed into outputs via intricate engineering, the latter are verified against user needs for accuracy, and all of these design control activities are maintained over a Design History File (DHF).
Medical Device Compliance
Addressing regulations and compliance checks forms the basis of medical device design engineering due to surging concerns around high health protection of patients and safety concerns. This majorly involves quality assurance and device efficacy proven through regulatory standards as and when it is introduced to the market. Businesses need to closely follow design guidelines like ISO 9001:1994 Quality-Systems Model etc. put forward by regulatory bodies such as International Electrotechnical Commission (IEC), US FDA’s Quality System Regulation (QSR), EU MDR which have been adapted across regions in different formats for marketing legally.
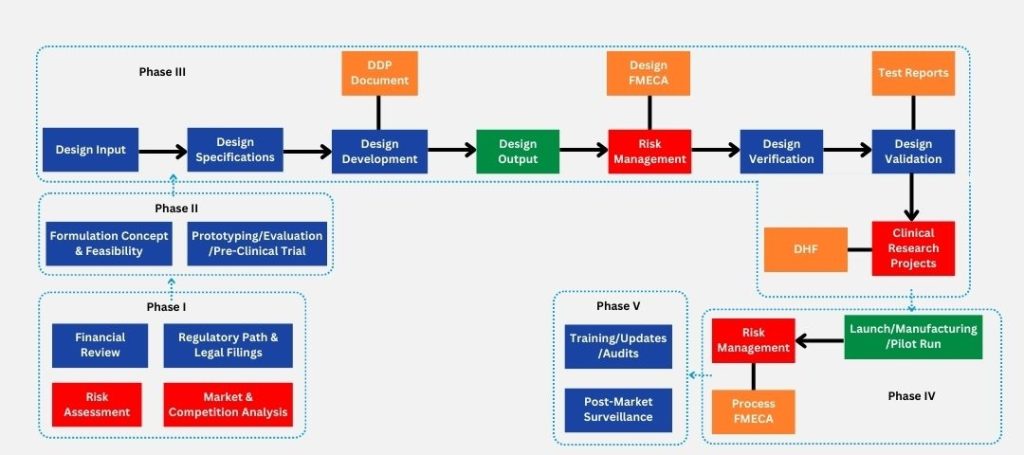
Brief outlay of medical device design process
Device Testing & Validation
As an integral part of medical device design services and engineering, testing and verification ensure that the medical device developed matches design inputs, features fixed specifications, customizations, user interface, intended functions etc. At the same time, medical device testing and validation ensures that it meets user requirements and evaluates its functionality or reliability from a real-world context to proceed with clinical trials. Businesses need to strategize on testing lab locations, test sequences and demonstrate compliance with standards and alignment with user needs.
Prior to launch, a quality management system placed ensures set of processes, responsibilities, quality objectives, and document management to meet quality targets, reduce waste, improve procedures, reduce costs, identify training opportunities, engage and direct resources, and ultimately satisfy customers.
Launch
This involves devising targeted geographies, meeting market standards, pricing, regulations, planning activities in a manner such that cost and time-to-market are optimized. Businesses must put in place risk management policies as a part of their medical device engineering services or minimum viable product vs prototype approach to ensure its usability. This process involves identifying hazards involving components, environment, their severity, mitigation and analysis such as Hazard and Operability Analysis (HAZOP), User-related Risk Analysis (URRA), Failure Mode Effects Analysis (FMEA) for expected failures.
Best Practices for Medical Device Design Services
Regulatory Compliances
As a key aspect of this segment, designers and developers of medical devices need to make sure that they adhere to regulations governing product safety and efficacy which may vary as per geography and respective bodies responsible for overseeing the process. For example, Food and Drug Administration is the regulatory authority in USA that classifies medical device design and development as per their risk category which includes –
1. Class I: These devices pose low risk to patients or users and do not require stringent pre-market approval, although they must comply with general controls such as proper labeling and manufacturing, for example, bandages, tongue depressors etc.
2. Class II: These devices pose moderate risks and require a 510(k) premarket approval in terms of their efficacy and safety, for example, infusion pumps, imaging, portable diagnostic devices, diagnostic and therapeutic equipment etc.
3. Class III: These are high risk category devices and must undergo a premarket approval process such as clinical trials before launching, for example, pacemakers, heart valves etc.
Medical Device Regulation (MDR 2017/7/45) established by the European Union sets out the standard requirements for placing medical devices in this region. It categorizes devices into Class I, IIa, IIb and III as per their risk level evaluated for compliance and CE-marked by a notified body, which is an accredited third-party organization. Similarly, regulatory agencies like Health Canada, PMDA Japan, TGA Australia etc. have their specific set of requirements for medical device design services and engineering solutions.
It therefore becomes necessary for manufacturers to be completely aware of the regulations of the geographical market that they are planning to sell the devices in. In addition to these, compliance with International Organization for Standardization (ISO 13485) and the IEC, which are internationally recognized quality management standard that describes development, production and maintenance of medical devices, is also important. These standards are also influenced by the International Medical Device Regulators Forum (IMDRF) which aims for an international convergence of regulations governing related practices. All of these platforms and organizations are directed under the authority of the World Health Organization (WHO) and United Nations that contribute to ISO and IEC as Category A liaison.
Process Outline
User Needs
As we discussed earlier, medical device design and development is broken down into several stages, each of which requires precise planning and execution. Businesses need to engage actively with healthcare professionals, patients and other stakeholders to understand user needs and gather their inputs. In-depth market research and clearly defining intended use of the device must be done for making development in par with performance, safety and usability. A user-centered approach can improve device interactions, reduce training requirements, errors, malfunctions in extreme conditions (temperature variations, chemical exposure, intensive use), increase efficiency, patient care quality and ensure seamless user journey. It also improves accessibility to users with disabilities, reduced mobility, limited vision, dexterity and adaptation to a broader range of lingual contexts.
Brainstorming session for conceptual designing, materials, functional requirements, budget constraints, aesthetic considerations (color, shape, size, texture, material choice), ergonomics (grip, weight, positioning) and ease of use must be considered prior to iterative designing, rapid prototyping such as 3D printing and fine-tuning of physical models and test concepts to foster innovation at every stage.
Design Control
Detailed product designing and engineering stage needs to be put in place once the concept is finalized. This includes finalizing specifications, medical prototype development, testing, material selection, and standard considerations. Associated risks must be identified through hazard analysis, severity assessment and measures for risk management in medical device industrial design as per ISO 14971 must be put in place including suggested design changes, time and costs involved in change-order, labelling, protective features etc. The devices’ safety and efficacy must be demonstrated using clinical trials, requisite data must be collected, and adequate testing needs to be conducted to ensure compliance with the regulatory standards.
Design Verification
The main processes involved in these include cost control, test period to reduce time-to-market, material evaluation for future variants, sequence testing etc. On clinical validation, medical device engineering services companies or manufacturers need to submit documentation of designs, processes, risk control strategies and clinical trial data under review for FDA-compliant medical devices 510(k) notifications or PMA submissions, HIPAA compliance, CE markings etc. as per target market. Post-approval, production quality control measures must comply with good manufacturing practices (GMP) standards. Continuous post-market surveillance and collection of user feedback is required to track device performance, adverse events, defects, implementation of device recalls etc. if necessary.
Quality Assurance
Real-world requirements are best understood when effective engagement with healthcare providers and patients is involved during early design process stages. All aspects of the device right from design to production process must meet high-quality standards through functional, safety, usability and clinical testing. Businesses must stay up to date with relevant regulations and regional medical device design engineering standards of the target market, while identifying, assessing, controlling and mitigating risks throughout the design and development process. Detailed records of all designs, testing and regulatory processes should be ensured for compliance and to support audits. It is necessary to assign ample time for each stage of the process for maximizing patient safety and complying with regulations to avoid market rejection. Post-launch, healthcare professionals and patients must be adequately trained in device usage, and its performance must be tracked for addressing any type of issue promptly.
Internet of Things or IoT in healthcare devices is another impactful segment that has contributed to enhancing the design and development of medical devices over the past few decades. It augments real-time data collection, remote patient monitoring, predictive analytics concerning the functional status of the device and empowers connected health devices (IoMT). Thus, improving patient outcomes, enabling personalized treatments, timely maintenance of smarter devices, and better decision-making in clinical environments.
Conclusion
The complex landscape of medical devices requires a multidisciplinary approach that addresses user needs and meets stringent regulations. Each phase, right from ideation to manufacturing, plays a pivotal role in the development of compliant and reliable medical devices. KritiKal solutions is one of the prominent embedded systems design companies in India, which strictly follows all requisite quality assurance procedures and standards such as ISO 13485 for design, development, manufacturing, installation and servicing. We focus on innovation, compliances and quality during new safe and reliable development, assurance of regulatory compliances, testing, design and risk controls. With KritiKal, businesses can master medical device development and contribute significantly to improving patient lives across the world. Please mail us at sales@kritikalsolutions.com to avail our medical device development services and widen the horizons of healthcare advancements.

Anmol Gupta currently works as an Embedded Firmware Engineer at KritiKal Solutions. He has over 3 years of experience in working on embedded systems, Firmware Designing, Analog and Digital semiconductors, Hardware analysis, PCB layout, PIC, ESP32, STM Microcontrollers, and has filed 5+ patents and published one research paper over the years. He is proficiently skilled in problem solving, Embedded C, C++, Python, CSS, HTML, Git, SQL, Arduino, MPLAB, STM etc. and has helped KritiKal in delivering some major projects with his design engineering skills.